
澳大利亞 TGA 認證澳大利亞的醫療器械分類規則幾乎與歐盟MDR分類相同,根據其風險等級分為
TGA注冊申請必須通過澳大利亞代理人才能完成
申請TGA時,為制造商做TGA注冊登記;
作為制造商和TGA之間的聯絡人,處理設備投訴、不良事件及召回等工作;根據TGA的要求提供有關設備的信息和文檔;

申請TGA時,為制造商做TGA注冊登記;
作為制造商和TGA之間的聯絡人,處理設備投訴、不良事件及召回等工作;根據TGA的要求提供有關設備的信息和文檔;

1、首先確定產品所屬類別;
2、指定巴西注冊持有人(BRH),該BRH必須獲得ANVISA認證的許可;
3、授權給該BRH,允許其代理申請ANVISA認證注冊并提交相關文件,以及代理BGMP審核申請
4、產品獲得INMETRO認證;產品必須通過ILAC成員實驗室的符合巴西標準要求的檢測,并獲得INMETRO授權機構簽發的INMETRO證書。證書有效期5年,每年通過驗廠維護證書的有效性;

注冊需要遵循醫療器械法規Medical Devices Regulations 2002?(SI 2002 No 618, as amended) (UK MDR 2002)。該法規基于歐盟的醫療器械法規MDD(93/42/EEC)、AIMDD(90/385/EEC)
和IVDD (98/79/EC)所制定,根據英國的情況做了適當調整。

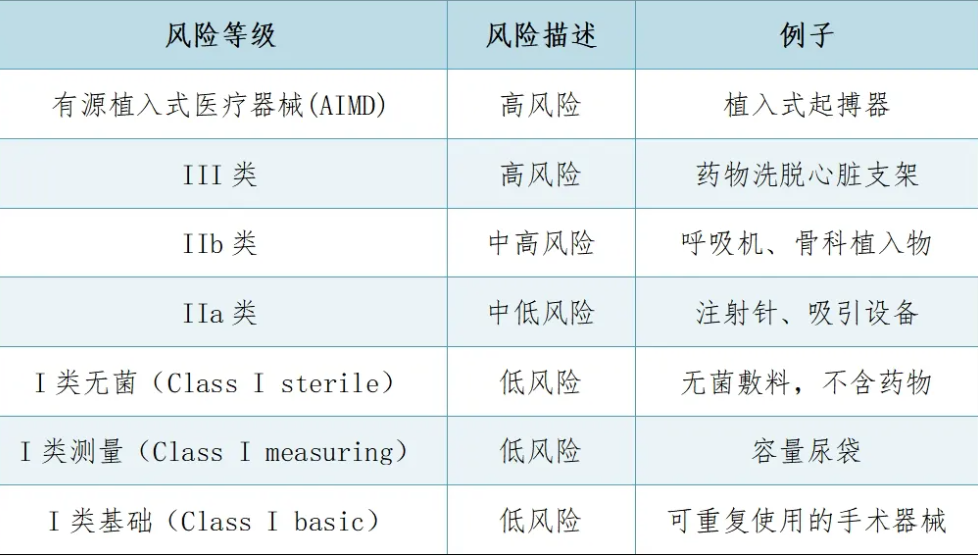
醫療器械和體外診斷產品根據其風險程度分為4類(A類、B類、C類、D類)。隨著器械類別從A類到D類,監管控制強度也逐步增加,A類器械受最少的監管控制(豁免注冊,列名即可),而D類器械則受到最嚴格的監管控制
某些醫療器械可能受其他法律影響,如:1992年電力法(Electricity Act 1992),1977年避孕、絕育和墮胎法(Contraception, Sterilisation and Abortion Act 1977),1996年危險物質和新生生物法(Hazardous Substances and New Organisms Act 1996),2016年輻射安全法(Radiation Safety Act 2016)

辦理流程及資料要求:確定產品分類→填寫“預先許可”申請表、附上產品外包裝樣稿及說明書 → 遞交“進口醫療衛材產品的預先許可申請”→預先許可申請通過→ 申請“產品進口準照”

根據 Conformity Assessment Procedures for Medical Device Approved by Recognised Countries: 已經在認可國家中獲得上市許可的產品,可以進行簡單產品CAB審核(證書+產品注冊文檔Verification驗證 + QMS審核的簡單CAB審核路徑,非全流程CAB審核)

境外生產商首先需要指定俄羅斯境內的代理持證人,角色包含總代、注冊代理等,直接與Roszdravnadzor溝通注冊等事宜。代理持證公司具體收費(代持證書費用、進口業務費用等)自定。










 客服1
客服1